Publication
Activation energies of sigmatropic shifts in propene and acetone enolate from the anti-Hermitian contracted Schrödinger equation
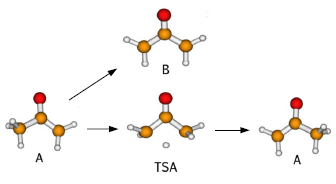
The hydrogen [1,3]-sigmatropic shift in propene is predicted by the Woodward–Hoffman rules to occur by an antarafacial pathway, yet the lack of experimental evidence suggests that this pathway is not favorable. Two natural questions arise: (i) can the [1,3]-shift be made more favorable by a symmetry-forbidden multistep pathway, and (ii) can the energetics be influenced by a substituent on propene? As in many chemical reactions, describing the energetics of these reactions requires a balanced treatment of both single-reference and multireference electron correlations, and yet traditional wave function methods often excel in treating only one kind of correlation. An equitable description of correlation effects, however, can be achieved, at a cost similar to efficient single-reference methods, by computing the two-electron reduced density matrix (2-RDM) from the anti-Hermitian part of the contracted Schrödinger equation (ACSE) [D. A. Mazziotti, Phys. Rev. Lett. 97, 143002 (2006)]. As with the contracted Schrödinger equation, the indeterminacy of the ACSE is removed without the many-electron wave function by reconstructing the 3-RDM from the 2-RDM via cumulant theory [D. A. Mazziotti, Chem. Phys. Lett. 289, 419 (1998)]. In this paper we apply the ACSE to study sigmatropic shifts in both propene and acetone enolate while extending its formalism to treat doublet spin states. In the 6-311G∗∗6-311G∗∗ basis set the ACSE predicts the activation energy of the trimethylene-to-propene rearrangement to be 8.8 kcal/mol while multireference perturbation theory yields a smaller barrier of 2.2 kcal/mol and coupled cluster singles-doubles predicts a negative barrier. We further find that the [1,3]-shift in acetone enolate is more favorable by ≈30kcal/mol≈30 kcal/mol than the [1,3]-shift in propene, which is consistent with a prior theoretical investigation as well as experimental observations of these shifts in 2-butanone enolate.